
- Case-Based Roundtable
- General Dermatology
- Eczema
- Chronic Hand Eczema
- Alopecia
- Aesthetics
- Vitiligo
- COVID-19
- Actinic Keratosis
- Precision Medicine and Biologics
- Rare Disease
- Wound Care
- Rosacea
- Psoriasis
- Psoriatic Arthritis
- Atopic Dermatitis
- Melasma
- NP and PA
- Skin Cancer
- Hidradenitis Suppurativa
- Drug Watch
- Pigmentary Disorders
- Acne
- Pediatric Dermatology
- Practice Management
- Prurigo Nodularis
- Buy-and-Bill
Publication
Article
Dermatology Times
Blau Syndrome: A Look Into a Rare Disease Requiring Collaborative Treatment
Author(s):
The autoinflammatory condition is associated with mutations in the NOD2 gene and usually affects children younger than 4 years.
Blau syndrome is a rare autosomal-dominant disease, affecting fewer than 1000 people total in the United States.1 The disease is characterized by a triad of dermatitis, arthritis, and uveitis.1 The autoinflammatory condition is associated with mutations in the NOD2 gene and usually affects children younger than4 years.1 Although Blau syndrome is predominantly hereditary, it has been described as having both an inherited/familial form and a sporadic form2 and may present differently in every patient.
Because Blau syndrome is a multisystem inflammatory condition, optimum disease management requires a collaborative effort among dermatologists, pediatricians, and ophthalmologists.3


Multiple, reddish-brown papules coalesced over the right arm in a boy with Blau syndrome. Photo courtesy: Donald A Glass II MD, PhD, Jennifer Maender MD, Denise Metry MD/Dermatology Online Journal
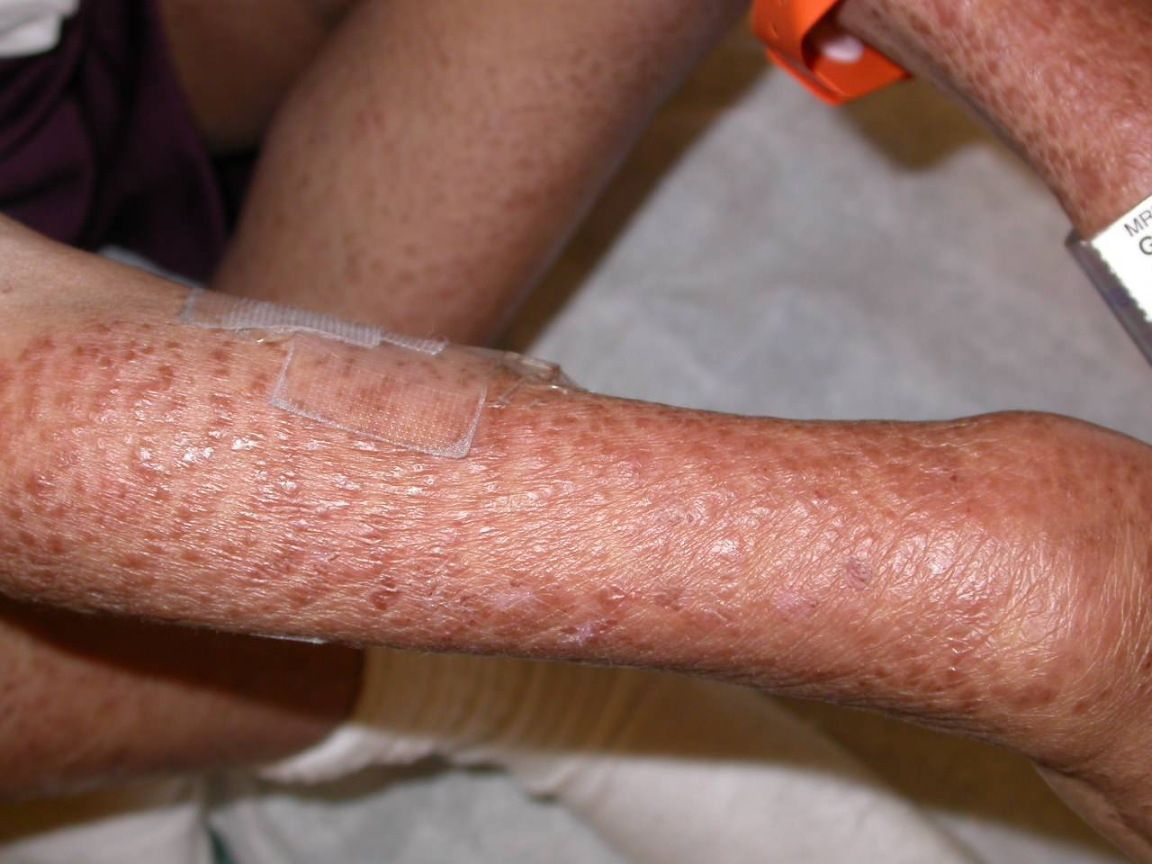
Clinical Presentation
Blau syndrome is associated with mutations in the NOD2 gene, which encodes the NOD2 protein. This protein is mainly expressed in macrophages, monocytes, and dendritic cells, playing an important role in innate immune defense against pathogens.4 Mutations in the NOD2 gene cause hyperactivation of nuclear factor–κB, resulting in an autoinflammatory disease with an exuberant response.2 The condition can be diagnosed through family history, clinical features, and noncaseating granulomas. The diagnosis can then be confirmed by genetic analysis.
Blau syndrome usually begins in early childhood, with skin rashes (most commonly appearing as brown-red papules) being the earliest sign. Another clinical manifestation includes arthritis that is usually symmetrical and polyarticular with marked tenosynovitis. The arthritis mainly involves wrists, knees, ankles, and proximal interphalangeal joints. Camptodactyly can later develop due to flexion contracture of proximal interphalangeal joints.2 In some cases, arthritis is often erosive and may cause joint deformity, which may lead to disability if not properly treated.5
Ocular manifestations usually develop later in the course of the disease; among them, the most common include recurrent bilateral uveitis. Other complications include conjunctival granulomas, keratoconjunctivitis sicca, optic nerve involvement, cataract, band-shaped keratopathy, and glaucoma. Uveitis is an important diagnostic indicator of Blau syndrome; however, it must be noted that 10% to 20% of patients with the disease may not develop ocular symptoms.5
Fever, large-vessel granulomatous vasculitis, malignant hypertension, and granulomatous inflammation of the kidneys, liver, and lungs are some other clinical manifestations of the disease.2 Besides classical clinical features, noncaseating epithelioid cell and giant cell granulomas in the affected tissues are the hallmark of the disease.
Treatment
Treating Blau syndrome is a challenging process. Collaborative efforts among dermatologists, pediatricians, and ophthalmologists are needed for optimum disease management of this multisystem inflammatory condition.3 Although Blau syndrome cannot be cured completely, several treatment strategies can help relieve symptoms associated with the condition.
Because mutations in a single gene are responsible for causing the disease, Blau syndrome is usually treated as a monogenic autoinflammatory disease. Treatment mainly focuses on preventing progressive ocular symptoms and resulting vision loss, as well as avoiding joint deformation.
The disease is usually treated with oral prednisolone alone or in combination with azathioprine, cyclosporine, methotrexate, or mycophenolate mofetil (CellCept). High-dose prednisolone can be used during flare-ups, and low doses help between the flare-ups. Tumor necrosis factor α inhibitors, including adalimumab(Humira) and infliximab (Remicade), can also be used.6 According to a study published in the Archives of Rheumatology, methotrexate and tumor necrosis factor inhibitors are more effective in patients with predominant articular symptoms.4 Moreover, IL-1β receptor antagonists have been reported to have variable outcomes.6
References
- Blau syndrome. Genetic and Rare Diseases Information Center. Accessed January 12, 2023.https://rarediseases.info.nih.gov/diseases/304/blau-syndrome
- Yi Yong C, Mukhtyar C, Armon K. 65. Blau syndrome treated with sequential biologics. Rheumatol Adv Pract. 2018;2(suppl 1):rky034.028. doi:10.1093/rap/rky034.028
- Agarwal A, Karande S. Blau syndrome: an under-reported condition in India? J Postgrad Med. 2022;68(2):63-67. doi:10.4103/jpgm.jpgm_1016_21
- PaÇKisaarslan A, SÖzerİ B, Şahİn N, et al. Blau syndrome and early-onset sarcoidosis: asix case series and review of the literature. Arch Rheumatol. 2019;35(1):117-127. doi:10.5606/ArchRheumatol.2020.7060
- Kumrah R, Pilania RK, Menia NK, et al. Blau syndrome: lessons learned in a tertiary care centre at Chandigarh, North India. Front Immunol. 2022;13:932919. doi:10.3389/fimmu.2022.932919
- Arvesen KB, Herlin T, Larsen DA, et al. Diagnosis and treatment of Blau syndrome/early-onset sarcoidosis, an autoinflammatory granulomatous disease, in an infant. Acta Derm Venereol. 2017;97(1):126-127. doi:10.2340/00015555-2485
























